Biokompatibilität setzt sich aus dem Zusammenspiel eines technischen Systems (das Medizinprodukt, das Bauteil, das Material) und eines biologischen Systems (der Patient, der Anwender) zusammen. Das technische System darf sich nicht negativ auf das biologische System auswirken. Ziel der Biokompatibilität ist der Schutz des Menschen vor biologischen Risiken, die sich aus der Anwendung eines Medizinprodukts ergeben.
Was besagt die EN ISO 10993-1?
Die EN ISO 10993-1 (Biologische Beurteilung von Medizinprodukten – Teil 1: Beurteilung und Prüfungen im Rahmen eines Risikomanagementsystems) – befasst sich mit der Beurteilung der Materialien des Medizinprodukts bzw. mit dem Medizinprodukt selbst.
Der Anwendungsbereich der Norm umfasst die Beurteilung der biologischen Sicherheit von aktiven, nicht aktiven, implantierbaren und nicht implantierbaren Medizinprodukten. Die Medizinprodukte werden nach Art und Dauer des Körperkontakts eingeteilt. Außerdem werden vorhandene Daten ausgewertet und falls nötig zusätzliche Datensätze ermittelt. Durch Veränderungen des Medizinprodukts im Laufe der Zeit (z.B. durch Wiederaufbereitungen oder Beschädigung des Medizinprodukts oder einzelner Bestandteile) kann der menschliche Körper neu entstandenen Materialien ausgesetzt sein. Auch diese biologische Gefährdung wird beurteilt. Durch eine Risikoanalyse werden die vorhandenen Datenlücken ermittelt.
Anhang A, Tabelle A.1
Die in einer biologischen Risikobewertung zu behandelnde Endpunkte der EN ISO 10993-1 zeigt die sogenannte "Endpunkttabelle". Aus Art und Dauer des Körperkontakts ergeben sich die verschiedenen Biokompatibilitätstests.
Die Norm legt dabei jedoch keine Teststrategie fest, da je nach Medizinprodukt auf einem risikobasierten Ansatz entschieden wird (EN ISO 10993-1:2020; Anhang A; A.1 Allgemeines): Die Endpunkttabelle soll keine fixe Checkliste sondern einen Rahmen für die Bewertung der Biokompatibilität und möglicher Prüfungen aufweisen. Angegebene relevante Endpunkte sind nicht zwangsläufig zu testen, jedoch sollten bestehende Datensätze bewertet werden, um mögliche fehlende Informationen zu identifizieren. Diese zusätzliche Identifizierung kann durch Erhebung zusätzlicher Datensätze oder durch Prüfungen erfolgen. Werden keine weiteren Datensätze erhoben, ist eine Begründung erforderlich.
EN ISO 10993-1:2020; Kapitel 5.2.2
Der Anwendungsbereich erfasst entweder den Patienten im Rahmen der bestimmungsgemäßen Verwendung mit dem Medizinprodukt oder den Anwender (z.B. bei OP-Handschuhen und Masken). Aber wie sieht es demnach mit Anwendern aus, die Kontakt zum Medizinprodukt oder Komponenten des Medizinprodukts haben, die nicht ausschließlich zum Schutz vorgesehen sind? Das regelt die Norm (EN ISO 10993-1:2020; Kapitel 5.2.2 Medizinprodukte mit Kontakt zu Körperoberflächen; a) Haut), sofern es ausschließlich um Hautkontakt geht, wie folgt:
- Ist zu belegen, dass das Material von Komponenten für andere Verbrauchsprodukte mit ähnlicher Kontaktart verwendet wird, braucht keine weitere biologische Beurteilung zu erfolgen
- Nicht durch Handschuhe geschützte Hände des Anwenders: Komponenten, die mit dem Anwender in Kontakt kommen können, z.B. menschliche Schnittstellen von elektronischen Geräten (Computertastaturen, Berührungsbildschirme) oder Gehäuse für elektronische Überwachungs- oder Programmiergeräte (z.B. Mobiltelefone, Tablets)
- Durch Handschuhe geschützte Hände des Anwenders: Komponenten, die mit dem Anwender in Kontakt kommen können (z. B. Kathetergriffe
Wie wird der Nachweis der biologischen Sicherheit praktisch umgesetzt?
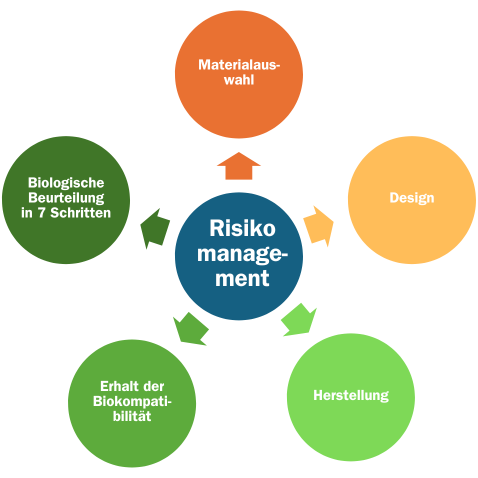
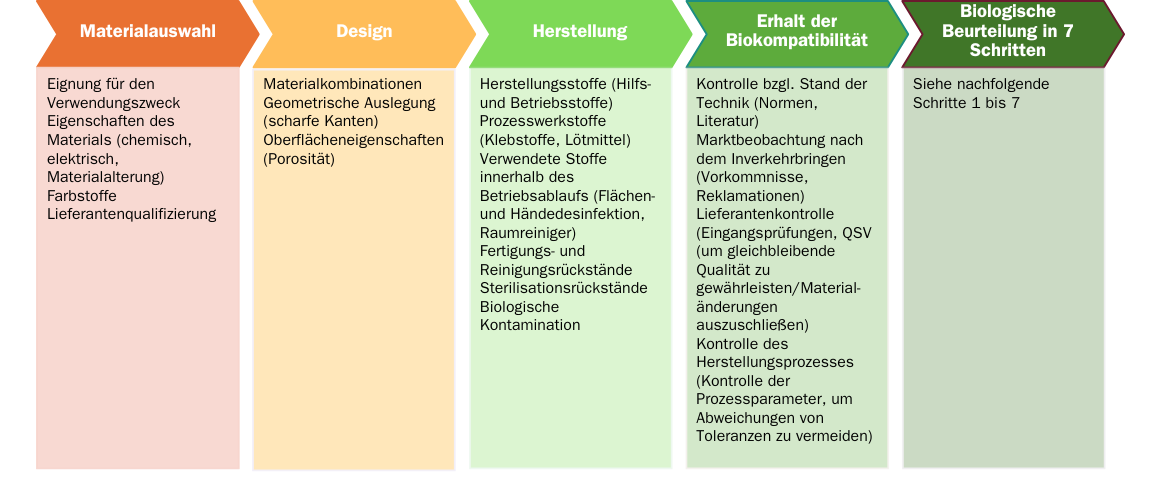
Die Biokompatibilität ist auf das Risikomanagement abzustimmen. Dabei sind folgende Punkte zu beachten: